DeepExplainer 基因组学示例
这使用在从 DeepLIFT 仓库(https://github.com/kundajelab/deeplift/blob/master/examples/genomics/genomics_simulation.ipynb)模拟的基因组数据上训练的模型运行 DeepExplainer,使用动态参考(即参考根据输入序列变化;在这种情况下,参考是输入序列的二核苷酸洗牌版本的集合)
模拟数据如下:
1/4 的序列同时包含 1-3 个 GATA_disc1 基序和 1-3 个 TAL1_known1 基序;这些被标记为 1,1,1
1/4 的序列包含嵌入的 GATA_disc1 基序,实例数为 1-3;这些被标记为 0,1,0
1/4 的序列包含 1-3 个嵌入的 TAL1_known1 基序;这些被标记为 0,0,1
1/4 的序列没有基序;这些被标记为 0,0,0
[1]:
%matplotlib inline
获取数据和Keras模型
拉入相关数据
[2]:
! [[ ! -f sequences.simdata.gz ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/db919b12f750e5844402153233249bb3d24e9e9a/deeplift/genomics/sequences.simdata.gz
! [[ ! -f keras2_conv1d_record_5_model_PQzyq_modelJson.json ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/b6e1d69/deeplift/genomics/keras2_conv1d_record_5_model_PQzyq_modelJson.json
! [[ ! -f keras2_conv1d_record_5_model_PQzyq_modelWeights.h5 ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/b6e1d69/deeplift/genomics/keras2_conv1d_record_5_model_PQzyq_modelWeights.h5
! [[ ! -f test.txt.gz ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/9aadb769735c60eb90f7d3d896632ac749a1bdd2/deeplift/genomics/test.txt.gz
加载数据
[3]:
! pip install simdna
Requirement already satisfied: simdna in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (0.4.3.2)
Requirement already satisfied: numpy>=1.9 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (1.26.3)
Requirement already satisfied: matplotlib in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (3.8.2)
Requirement already satisfied: scipy in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (1.11.4)
Requirement already satisfied: contourpy>=1.0.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (1.2.0)
Requirement already satisfied: cycler>=0.10 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (0.12.1)
Requirement already satisfied: fonttools>=4.22.0 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (4.47.2)
Requirement already satisfied: kiwisolver>=1.3.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (1.4.5)
Requirement already satisfied: packaging>=20.0 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (23.2)
Requirement already satisfied: pillow>=8 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (10.2.0)
Requirement already satisfied: pyparsing>=2.3.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (3.1.1)
Requirement already satisfied: python-dateutil>=2.7 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (2.8.2)
Requirement already satisfied: six>=1.5 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from python-dateutil>=2.7->matplotlib->simdna) (1.16.0)
[4]:
import gzip
import simdna.synthetic as synthetic
data_filename = "sequences.simdata.gz"
# read in the data in the testing set
test_ids_fh = gzip.open("test.txt.gz", "rb")
ids_to_load = [x.decode("utf-8").rstrip("\n") for x in test_ids_fh]
data = synthetic.read_simdata_file(data_filename, ids_to_load=ids_to_load)
[5]:
import numpy as np
# this is set up for 1d convolutions where examples
# have dimensions (len, num_channels)
# the channel axis is the axis for one-hot encoding.
def one_hot_encode_along_channel_axis(sequence):
to_return = np.zeros((len(sequence), 4), dtype=np.int8)
seq_to_one_hot_fill_in_array(
zeros_array=to_return, sequence=sequence, one_hot_axis=1
)
return to_return
def seq_to_one_hot_fill_in_array(zeros_array, sequence, one_hot_axis):
assert one_hot_axis == 0 or one_hot_axis == 1
if one_hot_axis == 0:
assert zeros_array.shape[1] == len(sequence)
elif one_hot_axis == 1:
assert zeros_array.shape[0] == len(sequence)
# will mutate zeros_array
for i, char in enumerate(sequence):
if char == "A" or char == "a":
char_idx = 0
elif char == "C" or char == "c":
char_idx = 1
elif char == "G" or char == "g":
char_idx = 2
elif char == "T" or char == "t":
char_idx = 3
elif char == "N" or char == "n":
continue # leave that pos as all 0's
else:
raise RuntimeError("Unsupported character: " + str(char))
if one_hot_axis == 0:
zeros_array[char_idx, i] = 1
elif one_hot_axis == 1:
zeros_array[i, char_idx] = 1
onehot_data = np.array(
[one_hot_encode_along_channel_axis(seq) for seq in data.sequences]
)
加载模型
[6]:
from keras.models import model_from_json
# load the keras model
keras_model_weights = "keras2_conv1d_record_5_model_PQzyq_modelWeights.h5"
keras_model_json = "keras2_conv1d_record_5_model_PQzyq_modelJson.json"
keras_model = model_from_json(open(keras_model_json).read())
keras_model.load_weights(keras_model_weights)
安装deeplift包以进行二核苷酸洗牌和可视化代码
[7]:
!pip install deeplift
Requirement already satisfied: deeplift in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (0.6.13.0)
Requirement already satisfied: numpy>=1.9 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from deeplift) (1.26.3)
计算重要性分数
定义生成引用的函数,在这种情况下,通过对给定的输入序列进行二核苷酸洗牌来实现。
[8]:
from deeplift.dinuc_shuffle import dinuc_shuffle
def shuffle_several_times(s):
s = np.squeeze(s)
return dinuc_shuffle(s, num_shufs=100)
使用动态引用函数运行 DeepExplainer
[9]:
import shap
np.random.seed(1)
seqs_to_explain = onehot_data[[0, 3, 9]] # these three are positive for task 0
dinuc_shuff_explainer = shap.DeepExplainer(
(keras_model.input, keras_model.output[:, 0]), shuffle_several_times
)
raw_shap_explanations = dinuc_shuff_explainer.shap_values(
seqs_to_explain, check_additivity=False
)
在单个序列上可视化分数
[10]:
from deeplift.visualization import viz_sequence
# project the importance at each position onto the base that's actually present
dinuc_shuff_explanations = (
np.sum(raw_shap_explanations, axis=-1)[:, :, None] * seqs_to_explain
)
for idx, dinuc_shuff_explanation in zip([0, 3, 9], dinuc_shuff_explanations):
print("Scores for example", idx)
highlight = {
"blue": [
(embedding.startPos, embedding.startPos + len(embedding.what))
for embedding in data.embeddings[idx]
if "GATA_disc1" in embedding.what.getDescription()
],
"green": [
(embedding.startPos, embedding.startPos + len(embedding.what))
for embedding in data.embeddings[idx]
if "TAL1_known1" in embedding.what.getDescription()
],
}
viz_sequence.plot_weights(
dinuc_shuff_explanation, subticks_frequency=20, highlight=highlight
)
Scores for example 0
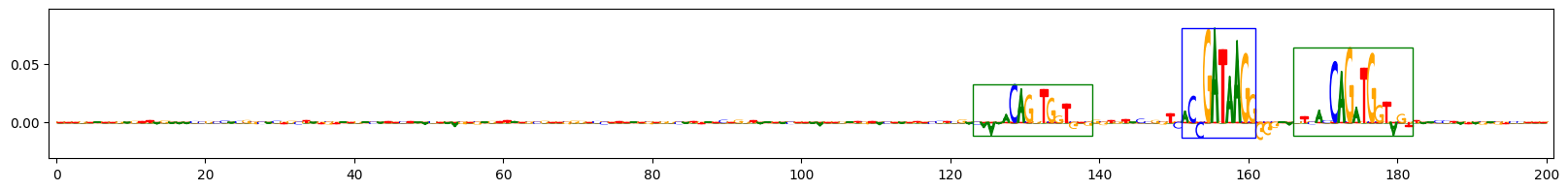
Scores for example 3
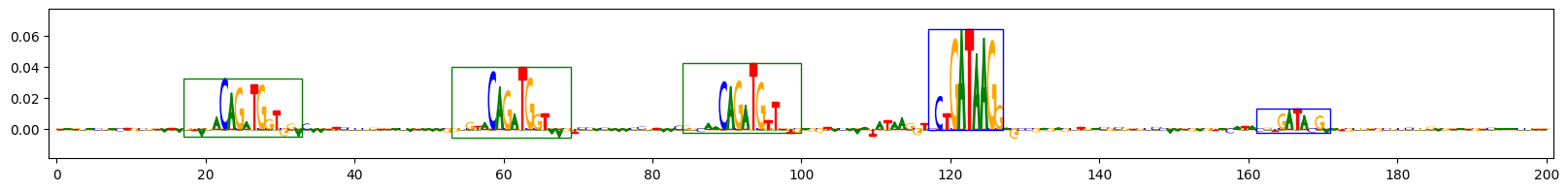
Scores for example 9
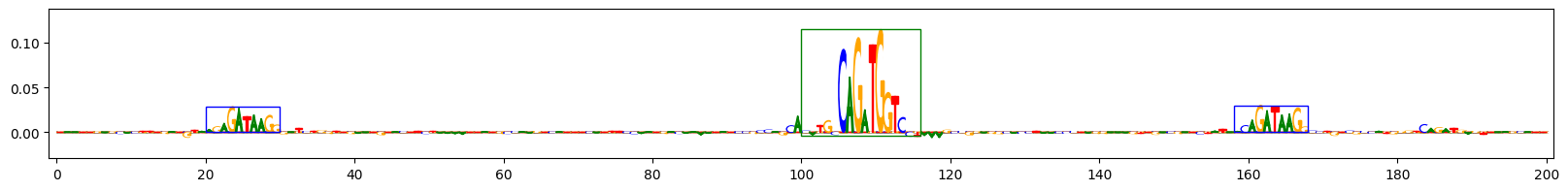
上面的图表展示了在预测同时包含GATA_disc1和TAL1_known1基序的序列任务中,三个示例序列的重要性得分。字母高度反映了得分。蓝色框表示插入的GATA_disc1基序的真实位置,绿色框表示插入的TAL1_known1基序的真实位置。